Duchenne muscular dystrophy (DMD) is a severe type of muscular dystrophy that primarily affects boys.[3] Muscle weakness usually begins around the age of four, and worsens quickly.[2] Muscle loss typically occurs first in the thighs and pelvis followed by the arms.[3] This can result in trouble standing up.[3] Most are unable to walk by the age of 12.[2] Affected muscles may look larger due to increased fat content.[3] Scoliosis is also common.[3] Some may have intellectual disability.[3] Females with a single copy of the defective gene may show mild symptoms.[3]
The disorder is X-linked recessive.[3] About two thirds of cases are inherited from a person's mother, while one third of cases are due to a new mutation.[3] It is caused by a mutation in the gene for the protein dystrophin.[3] Dystrophin is important to maintain the muscle fiber's cell membrane.[3] Genetic testing can often make the diagnosis at birth.[3] Those affected also have a high level of creatine kinase in their blood.[3]
Although there is no known cure, physical therapy, braces, and corrective surgery may help with some symptoms.[2] Assisted ventilation may be required in those with weakness of breathing muscles.[3] Medications used include steroids to slow muscle degeneration, anticonvulsants to control seizures and some muscle activity, and immunosuppressants to delay damage to dying muscle cells.[2] Gene therapy, as a treatment, is in the early stages of study in humans.[3] A small initial study using gene therapy has given some children improved muscle strength, but long term effects are unknown as of 2020.[6]
Various figures of the occurrence of DMD are reported. One source reports that it affects about one in 3,500 to 6,000 males at birth.[3] Another source reports DMD being a rare disease and having an occurrence of 7.1 per 100,000 male births.[7] A number of sources referenced in this article indicate an occurrence of 6 per 100,000.[8]
It is the most common type of muscular dystrophy.[3] The life expectancy is 26;[4] however, with excellent care, some may live into their 30s or 40s.[3] The disease is much more rare in girls, occurring approximately once in 50,000,000 live female births.[5]
Signs and symptoms
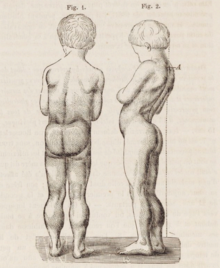
DMD causes progressive muscle weakness due to muscle fiber disarray, death, and replacement with connective tissue or fat.[3] The voluntary muscles are affected first, especially those of the hips, pelvic area, thighs, calves.[9][10] It eventually progresses to the shoulders and neck, followed by arms, respiratory muscles, and other areas.[10] Fatigue is common.[11]
Signs usually appear before age five, and may even be observed from the moment a boy takes his first steps.[12] There is general difficulty with motor skills, which can result in an awkward manner of walking, stepping, or running.[13] They tend to walk on their toes,[13] in part due to shortening of the Achilles tendon,[14] and because it compensates for knee extensor weakness.[10] Falls can be frequent.[15] It becomes harder and harder for the boy to walk; his ability to walk usually completely disintegrates before age 13.[13] Most men affected with DMD become essentially "paralyzed from the neck down" by the age of 21.[12] Cardiomyopathy, particularly dilated cardiomyopathy, is common, seen in half of 18-year-olds.[13] The development of congestive heart failure or arrhythmia (irregular heartbeat) is only occasional.[10] In late stages of the disease, respiratory impairment and swallowing impairment can occur, which can result in pneumonia.[16]
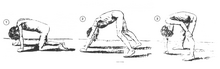
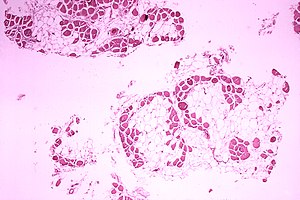
A classic sign of DMD is trouble getting up from lying or sitting position,[15] as manifested by a positive Gowers's sign. When a child tries to arise from lying on his stomach, he compensates for pelvic muscle weakness through use of the upper extremities:[13] first by rising to stand on his arms and knees, and then "walking" his hands up his legs to stand upright. Another characteristic sign of DMD is pseudohypertrophy (enlarging) of the muscles of the tongue, calves, buttocks, and shoulders (around age 4 or 5). The muscle tissue is eventually replaced by fat and connective tissue, hence the term pseudohypertrophy. Muscle fiber deformities and muscle contractures of Achilles tendon and hamstrings can occur, which impair functionality because the muscle fibers shorten and fibrose in connective tissue.[10] Skeletal deformities can occur, such as lumbar hyperlordosis, scoliosis, anterior pelvic tilt, and chest deformities. Lumbar hyperlordosis is thought to be compensatory mechanism in response to gluteal and quadriceps muscle weakness, all of which cause altered posture and gait (e.g.: restricted hip extension).[17][18]
Non musculoskeletal manifestations of DMD occur. There is a higher risk of neurobehavioral disorders (e.g., ADHD), learning disorders (dyslexia), and non-progressive weaknesses in specific cognitive skills (in particular short-term verbal memory),[13] which are believed to be the result of absent or dysfunctional dystrophin in the brain.[19]
Cause

DMD is caused by a mutation of the dystrophin gene, located on the short arm of the X chromosome (locus Xp21)[20] that codes for dystrophin protein. Mutations can either be inherited or occur spontaneously during germline transmission,[citation needed] causing to a large reduction or absence of dystrophin, a protein that provides structural integrity in muscle cells.[21] Dystrophin is responsible for connecting the actin cytoskeleton of each muscle fiber to the underlying basal lamina (extracellular matrix), through a protein complex containing many subunits. The absence of dystrophin permits excess calcium to penetrate the sarcolemma (the cell membrane).[22] Alterations in calcium and signalling pathways cause water to enter into the mitochondria, which then burst.[citation needed]
In skeletal muscle dystrophy, mitochondrial dysfunction gives rise to an amplification of stress-induced cytosolic calcium signals and an amplification of stress-induced reactive-oxygen species production. In a complex cascading process that involves several pathways and is not clearly understood, increased oxidative stress within the cell damages the sarcolemma and eventually results in the death of the cell. Muscle fibers undergo necrosis and are ultimately replaced with adipose and connective tissue.[citation needed]

DMD is inherited in an X-linked recessive pattern. Females typically are carriers of the genetic trait while males are affected. A female carrier will be unaware she carries a mutation until she has an affected son. The son of a carrier mother has a 50% chance of inheriting the defective gene from his mother. The daughter of a carrier mother has a 50% chance of being a carrier and a 50% chance of having two normal copies of the gene. In all cases, an unaffected father either passes a normal Y to his son or a normal X to his daughter. Female carriers of an X-linked recessive condition, such as DMD, can show symptoms depending on their pattern of X-inactivation.[citation needed]
DMD is extremely rare in females (about 1 in 50,000,000 female births).[5] It can occur in females with an affected father and a carrier mother, in those who are missing an X chromosome, or those who have an inactivated X chromosome (the most common of the rare reasons).[23] The daughter of a carrier mother and an affected father will be affected or a carrier with equal probability, as she will always inherit the affected X-chromosome from her father and has a 50% chance of also inheriting the affected X-chromosome from her mother.[24]
Disruption of the blood-brain barrier has been seen to be a noted feature in the development of DMD.[25]
Diagnosis
Genetic counseling is advised for people with a family history of the disorder. DMD can be detected with about 95% accuracy by genetic studies performed during pregnancy.[16] Creatine kinase (CPK-MM) levels in the bloodstream are extremely high. An electromyography (EMG) shows that weakness is caused by destruction of muscle tissue rather than by damage to nerves.[citation needed]
DNA test
The muscle-specific isoform of the dystrophin gene is composed of 79 exons, and DNA testing (blood test) and analysis can usually identify the specific type of mutation of the exon or exons that are affected. DNA testing confirms the diagnosis in most cases.[26]
Muscle biopsy
If DNA testing fails to find the mutation, a muscle biopsy test may be performed.[27] A small sample of muscle tissue is extracted using a biopsy needle. The key tests performed on the biopsy sample for DMD are immunohistochemistry, immunocytochemistry, and immunoblotting for dystrophin, and should be interpreted by an experienced neuromuscular pathologist.[28] These tests provide information on the presence or absence of the protein. Absence of the protein is a positive test for DMD. Where dystrophin is present, the tests indicate the amount and molecular size of dystrophin, helping to distinguish DMD from milder dystrophinopathy phenotypes.[29] Over the past several years, DNA tests have been developed that detect more of the many mutations that cause the condition, and muscle biopsy is not required as often to confirm the presence of DMD.[30]
Prenatal tests
A prenatal test can be considered when the mother is a known or suspected carrier.[31]
Prenatal tests can tell whether the unborn child has one of the most common mutations. Many mutations are responsible for DMD, and some have not been identified, so genetic testing may be falsely negative if the suspected mutation in the mother has not been identified.[citation needed]
Prior to invasive testing, determination of the fetal sex is important; while males are sometimes affected by this X-linked disease, female DMD is extremely rare. This can be achieved by ultrasound scan at 16 weeks or more recently by free fetal DNA (cffDNA) testing. Chorion villus sampling (CVS) can be done at 11–14 weeks, and has a 1% risk of miscarriage. Amniocentesis can be done after 15 weeks, and has a 0.5% risk of miscarriage. Non invasive prenatal testing can be done around 10–12 weeks.[32] Another option in the case of unclear genetic test results is fetal muscle biopsy.
Treatment

No cure for DMD is known, and an ongoing medical need has been recognized by regulatory authorities.[33] Gene therapy has shown some success.[34]
Treatment is generally aimed at controlling symptoms to maximize the quality of life which can be measured using specific questionnaires,[35] and include:
- Corticosteroids such as prednisolone and deflazacort lead to short-term improvements in muscle strength and function up to 2 years.[36] Corticosteroids have also been reported to help prolong walking, though the evidence for this is not robust.[37]
- Randomised control trials have shown that β2 agonists increase muscle strength, but do not modify disease progression. Follow-up time for most RCTs on β2 agonists is only around 12 months, hence results cannot be extrapolated beyond that time frame.[citation needed]
- Mild, nonjarring physical activity such as swimming is encouraged. Inactivity (such as bed rest) can worsen the muscle disease.
- Physical therapy is helpful to maintain muscle strength, flexibility, and function.
- Orthopedic appliances (such as braces and wheelchairs) may improve mobility and the ability for self-care. Form-fitting removable leg braces that hold the ankle in place during sleep can defer the onset of contractures.
- Appropriate respiratory support as the disease progresses is important.
- Cardiac problems may require a pacemaker.[38]
The medication eteplirsen, a Morpholino antisense oligo, has been approved in the United States for the treatment of mutations amenable to dystrophin exon 51 skipping. The US approval has been controversial[39] as eteplirsen failed to establish a clinical benefit;[40] it has been refused approval by the European Medicines Agency.[41]
The medication ataluren (Translarna) is approved use in the European Union.[42][43]
The antisense oligonucleotide golodirsen (Vyondys 53) was approved for medical use in the United States in 2019, for the treatment of cases that can benefit from skipping exon 53 of the dystrophin transcript.[44][45]
The Morpholino antisense oligonucleotide viltolarsen (Viltepso) was approved for medical use in the United States in August 2020, for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping.[46] It is the second approved targeted treatment for people with this type of mutation in the United States.[46] Approximately 8% of people with DMD have a mutation that is amenable to exon 53 skipping.[46]
Casimersen was approved for medical use in the United States in February 2021,[47] and it is the first FDA-approved targeted treatment for people who have a confirmed mutation of the DMD gene that is amenable to exon 45 skipping.[47]
Comprehensive multidisciplinary care guidelines for DMD have been developed by the Centers for Disease Control and Prevention, and were published in two parts in The Lancet Neurology in 2010.[27] An update was published in 2018.[48][49]
Physical therapy
Physical therapists are concerned with enabling patients to reach their maximum physical potential. Their aim is to:[50]
- minimize the development of contractures and deformity by developing a programme of stretches and exercises where appropriate
- anticipate and minimize other secondary complications of a physical nature by recommending bracing and durable medical equipment[51]
- monitor respiratory function and advise on techniques to assist with breathing exercises and methods of clearing secretions[50]
Respiration assistance
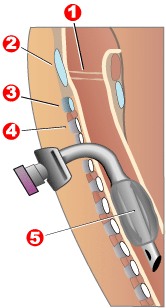
1 – Vocal folds
2 – Thyroid cartilage
3 – Cricoid cartilage
4 – Tracheal rings
5 – Balloon cuff
Modern "volume ventilators/respirators", which deliver an adjustable volume (amount) of air to the person with each breath, are valuable in the treatment of people with muscular dystrophy-related respiratory problems. The ventilator may require an invasive endotracheal or tracheotomy tube through which air is directly delivered, but for some people, noninvasive delivery through a face mask or mouthpiece is sufficient. Positive airway pressure machines, particularly bilevel ones, are sometimes used in this latter way. The respiratory equipment may easily fit on a ventilator tray on the bottom or back of a power wheelchair with an external battery for portability.[citation needed]
Ventilator treatment may start in the mid- to late teens when the respiratory muscles can begin to collapse. If the vital capacity has dropped below 40% of normal, a volume ventilator/respirator may be used during sleeping hours, a time when the person is most likely to be underventilating (hypoventilating). Hypoventilation during sleep is determined by a thorough history of sleep disorder with an oximetry study and a capillary blood gas (see pulmonary function testing).[citation needed] A cough assist device can help with excess mucus in lungs by hyperinflation of the lungs with positive air pressure, then negative pressure to get the mucus up. If the vital capacity continues to decline to less than 30 percent of normal, a volume ventilator/respirator may also be needed during the day for more assistance. The person gradually will increase the amount of time using the ventilator/respirator during the day as needed. However, there are also people with the disease in their 20s who have no need for a ventilator.[citation needed]
Future developments
There is no cure for any of the muscular dystrophies.[52] Several drugs designed to address the root cause are under development, including gene therapy (Microdystrophin), and antisense drugs (Ataluren, Eteplirsen etc.).[53] Other medications used include corticosteroids (Deflazacort), calcium channel blockers (Diltiazem) to slow skeletal and cardiac muscle degeneration, anticonvulsants to control seizures and some muscle activity, and immunosuppressants (Vamorolone) to delay damage to dying muscle cells.[2] Physical therapy, braces, and corrective surgery may help with some symptoms[2] while assisted ventilation may be required in those with weakness of breathing muscles.[3] Outcomes depend on the specific type of disorder.[54][53]
A paper published by Stanford University on 10 March 2022 demonstrated that patients with muscular dystrophies could benefit from new therapies targeting the specific pathways contributing directly to muscle disorders. Three recent advances are likely to enhance the landscape of treatments for muscular dystrophies such as DMD. First, induced pluripotent stem cells (iPSCs) allow researchers to design effective treatment strategies. Second, artificial intelligence (AI) can help identify therapeutic targets. Third, a high volume of multi-omics data gathered from diverse sources through disease models can provide valuable information about converging and diverging pathways.[21]
Prognosis
Duchenne muscular dystrophy is a rare progressive disease which eventually affects all voluntary muscles and involves the heart and breathing muscles in later stages. Life expectancy is estimated to be around 25–26,[16][4] but this varies. With excellent medical care, affected men often live into their 30s.[55] David Hatch of Paris, Maine, may be the oldest person in the world with the disease; as of 2021, he was 58.[56]
The most common direct cause of death in people with DMD is respiratory failure. Complications from treatment, such as mechanical ventilation and tracheotomy procedures, are also a concern. The next leading cause of death is cardiac-related conditions such as heart failure brought on by dilated cardiomyopathy. With respiratory assistance, the median survival age can reach up to 40. In rare cases, people with DMD have been seen to survive into their forties or early fifties, with proper positioning in wheelchairs and beds, and the use of ventilator support (via tracheostomy or mouthpiece), airway clearance, and heart medications.[57] Early planning of the required supports for later-life care has shown greater longevity for people with DMD.[58]
Curiously, in the mdx mouse model of Duchenne muscular dystrophy, the lack of dystrophin is associated with increased calcium levels and skeletal muscle myonecrosis. The intrinsic laryngeal muscles (ILMs) are protected and do not undergo myonecrosis.[59] ILMs have a calcium regulation system profile suggestive of a better ability to handle calcium changes in comparison to other muscles, and this may provide a mechanistic insight for their unique pathophysiological properties.[60] The ILM may facilitate the development of novel strategies for the prevention and treatment of muscle wasting in a variety of clinical scenarios.[61] In addition, patients with Duchenne muscular dystrophy also have elevated plasma lipoprotein levels, implying a primary state of dyslipidemia in patients.[62]
Epidemiology
DMD is the most common type of muscular dystrophy; it affects about one in 5,000 males at birth.[3] DMD has an incidence of one in 3,600 male infants.[16]
In the US, a 2010 study showed a higher amount of those with DMD age ranging from 5 to 54 who are Hispanic compared to non-Hispanic Whites, and non-Hispanic Blacks.[63]
History

The disease was first described by the Neapolitan physician Giovanni Semmola in 1834 and Gaetano Conte in 1836.[64][65][66] However, DMD is named after the French neurologist Guillaume-Benjamin-Amand Duchenne (1806–1875), who in the 1861 edition of his book Paraplegie hypertrophique de l'enfance de cause cerebrale, described and detailed the case of a boy who had this condition. A year later, he presented photos of his patient in his Album de photographies pathologiques. In 1868, he gave an account of 13 other affected children. Duchenne was the first to do a biopsy to obtain tissue from a living patient for microscopic examination.[67][68]